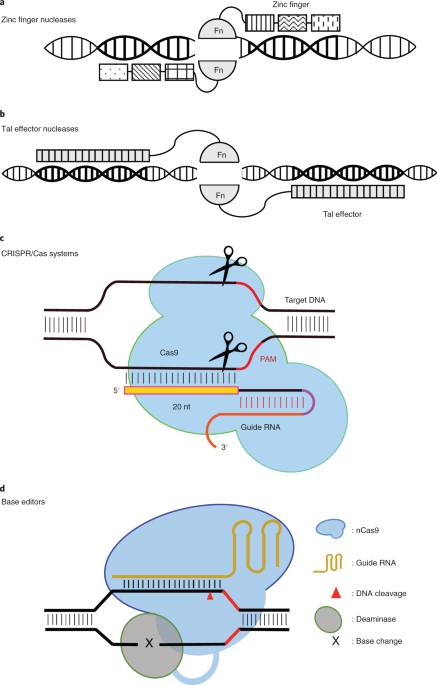
Genome editing using programmable nucleases, such as Zinc Finger Nucleases (ZFNs), Transcription Activator-like Effector Nucleases (TALENs) and CRISPR/Cas9 systems, is revolutionizing life sciences and medicine 1. Although programmable nucleases allow for site-specific and permanent alterations to the genomes of a wide variety of organisms, their off-target editing remains a significant concern, especially in therapeutic applications2, 3.
To address this challenge, both in silico and experimental tools for genome-wide off-target site prediction have been developed over the last few years, which have generated remarkably concordant results for sites with high off-target editing activity. However, no single tool is able to accurately predict low-frequency off-target editing, presenting a bottleneck in therapeutic genome editing, since even a small number of cells with off-target editing can be detrimental. Specifically, the current in silico prediction methods have been trained on a fairly limited set of genome-wide activity data and thus may not accurately rank off-target, and the existing experimental methods, such as GUIDE-seq and CIRCLE-seq4, 5, have their limitations. Clear guidance for identifying and validating highly specific genome editing target sites has not been established.
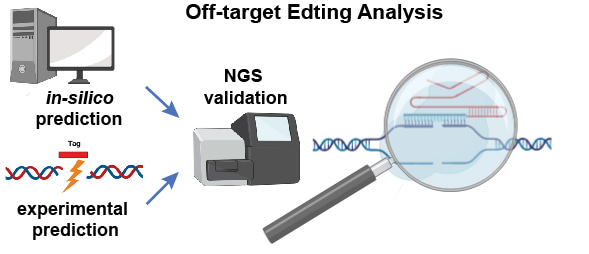
We therefore performed a critical review and analysis of the tools developed and compare their abilities to predict bona fide off-target sites. Based on our analysis, we recommend that at least one in silico tool and one experimental tool should be used together to identify potential off-target sites, and amplicon-based next-generation sequencing (NGS) should be used as the gold-standard assay for assessing the true off-target effects at these candidate sites (Figure 1).
Future work to improve off-target analysis includes the generation of a much larger dataset of true off-target sites, improving NGS techniques to increase sensitivity, and using personalized genomes instead of a common reference genome. In particular, an enlarged off-target dataset encompassing several dozen sgRNAs in several different cell types, which is feasible to obtain using current technology, will greatly aid in the development of this nascent field. It can be used as a gold standard to evaluate new experimental techniques; as a training dataset for machine learning approaches to off-target prediction; and as a starting point for analyzing the effect of chromatin structure and other aspects of the cell state on off-target gene editing. These analyses will help define the best practices and serve as a roadmap towards standardizing the off-target analysis for nuclease-based genome editing.
Our paper: Bao et al. Tools for Experimental and Computational Analyses of Off-target Editing by Programmable Nucleases, Nature Protocols (2020).

Figure 1. A schematic illustration of off-target analysis for programmable nuclease based genome editing. Both in silico and experimental prediction tools should be used to identify potential off-target sites, which are then validated using amplicon-based next-generation sequencing (NGS).
Reference
- Cox, D.B., Platt, R.J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat Med 21, 121-131 (2015).
- Hsu, P.D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827-832 (2013).
- Cradick, T.J., Fine, E.J., Antico, C.J. & Bao, G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41, 9584-9592 (2013).
- Tsai, S.Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology (2015).
- Tsai, S.Q. et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607-614 (2017).





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in