
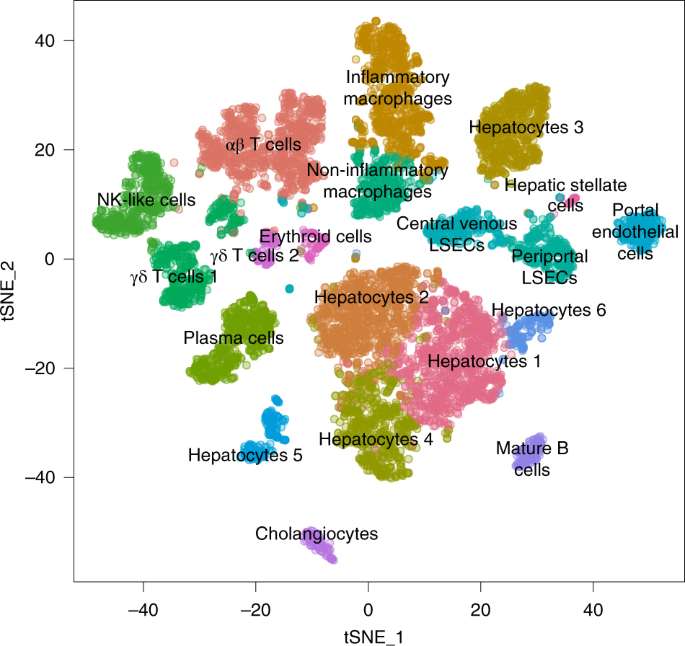
How does the body work? As technology has evolved, we have been able to answer this question with increasing resolution and precision. Functions were first assigned to organs, then tissues, and eventually cells, and even the molecules within cells. Now, with single-cell RNA-sequencing (scRNA-seq), we are able to sequence the RNA that is being produced in thousands of cells simultaneously in a single experiment. Because it does not depend on prior expectations of cell identity, single-cell transcriptomics has enabled the discovery of new cell types and has revolutionized our understanding of healthy and diseased tissue. It is increasingly clear that tissues are complex ecosystems of cells that grow, develop, and interact with each other. This is even thought to be true in diseases such as cancer, which has proven to be much more heterogeneous than what has often been thought of as a single, rapidly proliferating cell clone. Single-cell transcriptomic maps are used to visualize these cellular ecosystems to support interpretation. On one of these maps, each dot represents an individual cell from the experiment, and cells are grouped together based on the similarity between their transcriptomes. This visualization is performed with computational tools that can reduce the high dimensional gene expression matrix of a cell down to two coordinates that can be placed on a map that we can see. The different clusters are then labeled with their hypothesized cell-type labels derived from interpreting the genes that are expressed by individual cells, or averaged across clusters of similar cells.
With thousands of cells often measured for one sample, and with increasing numbers of samples analyzed, the process of labeling a single-cell map needs to be efficient and accurate. One challenge is that there is considerable variability in tissue types, experimental protocols and measured gene expression patterns, and it is not always clear where to start and where to stop in the annotation task. To address this, we have created a tutorial that explains how to interpret and label a cellular map derived from scRNA-seq data from start to finish. We recommend following three steps for single-cell map annotation: automatic cell annotation, manual cell annotation and verification. We also provide guidance about decision making that we hope is useful to those who are both new to, and already familiar with, analysing and interpreting scRNA-seq data.
As we formulated a procedure to improve the consistency of the annotation process, we found that a key challenge was balancing accuracy with speed. Clusters can be labeled using well-known marker genes to manually pick out major cell types.While often very quick to start for biological experts, this approach usually ends up being more time consuming than expected, as the annotation process needs to be repeated many times while optimizing clustering and quality control parameters, as well as redone every time a new sample is added to a data set. A solution to this is to use automatic annotation tools to assign cell-type labels to cells or clusters. However, improved speed compromises accuracy in this case, and manual work is still required to collect markers used as input to automated methods, confirm automatically predicted labels and annotate cells missed by automatic methods, to maintain a high quality map.
Part of what complicates labeling single-cell transcriptomic maps comes from the assumption made by clustering and annotation tools that cell types are discrete entities. It is more likely that cells exist on a continuum of types and states, and may be composed of overlapping states (e.g. inflamed, hypoxic and undergoing cell division, each following its own continuum). Because automatic cell-type annotation also relies on a reference data set of cells labeled with their corresponding genes, it also assumes we’ve already seen all of the cells in the sample before. Cells that defy these assumptions (e.g. cells that are in a transitional/developmental state, or in previously unrecognized states) are more difficult to automatically label and must often be manually scrutinized. In addition, it is not uncommon for clusters of cells to present unclear gene expression profiles due technical effects, which again emphasizes the challenges of automatic annotation.
scRNA-seq can identify new cell types and transitional or developmental states, especially when exploring a new tissue, species, disease state, or using new experimental protocols that may enhance the survival of sensitive cells. However, before claiming a novel cell type or transitional/developmental relationship between cells, it is important to rule out any technical reasons for these differences. An ambiguous group of cells could be the result of technical artifacts: multiple cells could be captured and sequenced as if they are an individual cell, creating “doublets”; groups of cells with particularly low numbers of RNA sequenced can be artificially clustered together; or cells sequenced at difference facilities or from different individuals can create “technical batch effects”, causing technical variability that can obscure true biological variability. All of these potential factors may influence cell labels and should be explored using quality control methods upstream of labeling cells or cell clusters.
We also include tutorial sections on data integration and cross-species comparisons. As single cell genomics becomes increasingly affordable and accessible, data will be generated from more model systems and these will be integrated or compared to better identify biological signals in the data. Good data integration that retains important biological signals while correcting for batch effects remains an important challenge, but, fortunately, multiple existing methods will usually work reasonably well at this task.
Finally, we emphasize that the cell labels we identify are hypotheses, and experimental tests are required to confirm the expression of marker genes, cellular function and the presence of specific cells and markers within a tissue sample. We hope our tutorial will be of broad use to the community and are always interested in getting feedback about our recommendations.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in